

近几年来医疗行业发展迅速,并且随着市场的需求不断增加,医疗器械生产及销售公司的数量也呈增加趋势。医疗器械作为医疗范畴内的行业分支,有所不同,它重点面向医院、基层医疗机构、体检中心等,我国医疗器械分类是依照风险水平由低到高划分为三个级别,其安全性能不同所需的管理权限不同。
第一类医疗器械是风险程度低、实行常规管理可以保证其安全有效的医疗器械,比如手术刀、手术剪、手动病床、医用冰袋、降温贴等,其产品和生产活动由所在地设区的市级食品药品监管部门实行备案管理。经营活动则全部放开,既不用许可也不用备案,只需取得工商部门核发的营业执照即可。
第二类医疗器械是具有中度风险,需要严格控制管理以保证其安全有效的医疗器械,比如我们日常生活中常见的创可贴、体温计、血压计、制氧机、雾化器等,其产品和生产活动由省级食品药品监管部门实行许可管理,分别发给《医疗器械注册证》和《医疗器械生产许可证》。经营活动由设区的市级食品药品监管部门实行备案管理;
第三类医疗器械是具有较高风险,需要采取特别措施严格控制管理以保证其安全有效的医疗器械,比如常见的输液器、注射器、静脉留置针、心脏支架、呼吸机、CT、核磁共振等,其产品和生产经营活动分别由国家总局、省级食品药品监管部门和设区的市食品药品监管部门实行许可管理,分别发给《医疗器械注册证》、《医疗器械生产许可证》、《医疗器械经营许可证》。
随着我国相关行业的技术革新及产业链的日渐成熟,医疗器械产业正在进入高速发展期,市场容量不断扩大,产品认证需求与日俱增。
一、什么是医疗器械注册/认证?
医疗器械注册/认证是食品药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。
医疗器械备案是医疗器械备案人向食品药品监督管理部门提交备案资料,食品药品监督管理部门对提交的备案资料存档备查。
医疗器械产品注册证是医疗器械产品上市销售的合格证明。
二、哪些产品需要进行医疗器械注册/认证?
被定义为医疗器械的产品需要进行注册/认证,这些产品通常是指制造商的预期用途是为下列一个或多个特定医疗目的用于人类的,不论单独使用或组合使用的仪器、设备、器具、机器、用具、植入物、体外试剂、软件或其他相似或相关物品。这些目的包括:
- 疾病的诊断、预防、监护、治疗或者缓解;
- 损伤的诊断、监护、治疗、缓解或者补偿;
- 解剖或生理过程的研究、替代、调节或者支持;
- 支持或维持生命;
- 妊娠控制;
- 医疗器械的消毒;
- 通过对取自人体的样本进行体外检查的方式来提供医疗信息; 其作用于人体体表或体内的主要设计作用不是用药理学、免疫学或代谢的手段获得,但可能有这些手段参与并起一定辅助作用。
值得注意的是,有些产品在某些管辖范围内可认为是医疗器械,而在其他地方不认为是医疗器械的产品包括:
- 消毒物质;
- 残疾人的辅助用品;
- 含有动物和(或)人体组织的器械;
- 用于体外受精或生育辅助的器械。
三、对于医疗器械注册/认证,当前国际国内的主流法规有哪些?
当前国际国内的主流法规有三大类,分别是中国NMPA医疗器械注册、欧盟CE认证和美国FDA认证:
1、 中国NMPA医疗器械注册/备案
根据中国国务院《医疗器械监督管理条例》规定,任何生产企业希望在中国境内销售、使医疗器械( 包含境内和境外的器械),都应当向相应的食品药品监督管理部门进行注册,按照其风险等级主要分为三类:
- 第一类医疗器械(风险程度低,如纱布、绷带等),实行产品备案管理;
- 第二类医疗器械(中度风险,如输液器、手术手套等),实行产品注册管理;
- 第三类医疗器械(较高风险,如支架、人工关节等),实行产品注册管理。
NMPA注册流程如下:
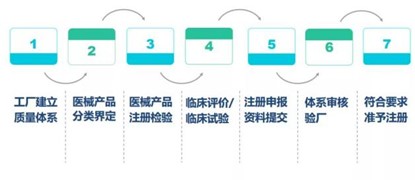
2、 欧盟CE认证
在欧盟市场,“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》法规的基本要求。欧盟CE认证是进入欧盟市场的通行证,出口到欧盟的医疗器械没有CE认证无法清关。医疗器械需要满足的CE指令有《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、《医疗器械指令》(MDD,93/42/EEC)和《体外诊断器械指令》(IVDD, 98/79/EC),根据医疗器械的风险不同,在欧盟市场医疗器械被划分为Ⅰ、Ⅱa、Ⅱb、Ⅲ四个类别:
- 低风险性医疗器械属于I类,包括:
普通I类医疗器械,需出具CE符合性报告;
具有无菌及测量功能的特殊I类医疗器械,需要CE证书,并在产品包装上打上CE标识。
- 中度风险性医疗器械属于IIa类和IIb类,需要CE证书,并在产品包装上打上CE标识。
- 高度风险性医疗器械属于III类,需要CE证书,并在产品包装上打上CE标识。
CE认证流程如下: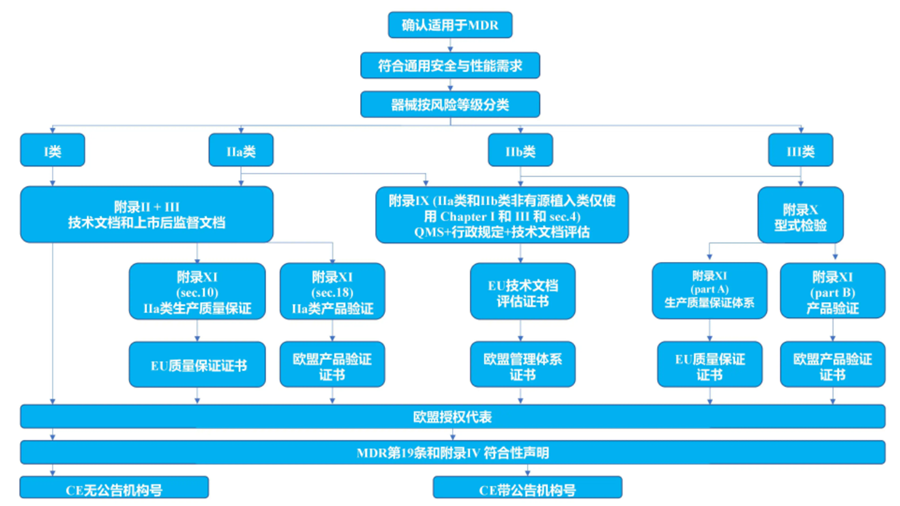
3、 美国FDA认证
FDA认证即美国食品药品监督管理局(Foodand Drug Administration,简称FDA),为确保美国本国生产或进口的食品、化妆品、药物、生物制剂、设备和放射产品的安全而设立的审查机制,在美国等近百个国家,只有通过了FDA认可的材料、器械和技术才能进行商业化临床应用。根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),其中Ⅲ类风险等级最高,风险等级越高则监督越多。FDA明确规定了每一种医疗器械的产品分类和管理要求,FDA医疗器械产品目录已收录超过1700多种产品。医疗器械FDA注册类型包括:
- 厂家在FDA注册;
- 产品的FDA登记;
- 产品上市登记(510表登记);
- 产品上市审核批准(PMA审核);
- 医疗保健器械的标签与技术改造、通关、登记、上市前报告。
FDA认证流程如下: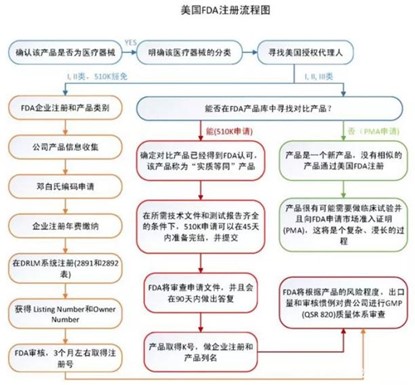
四、医疗认证过程中有什么难点和注意事项?
一般来说,医疗器械认证过程中有以下几点需要注意:
一是分析医疗器械特点,根据医疗器械的定义及风险等级,结合产品的预期用途、结构组成及产品描述,来综合判定产品的类别;
二是根据设计开发的输入阶段产品法规、标准的收集,确定产品技术要求中的性能指标的检测依据;
三是根据产品的分类来选择医疗器械认证过程中符合性评价程序和注册流程;
四是根据产品的预期用途及预期使用用途、适用人群及产品禁忌症和不良反应等信息,来决策是否需要进行临床评价,选择合适的临床评价路径;
本期就给大家介绍到这,想了解更多详细信息请关注大大通“一条胖头鱼”!
本文参考资料来自华通威检测认证机构。
评论